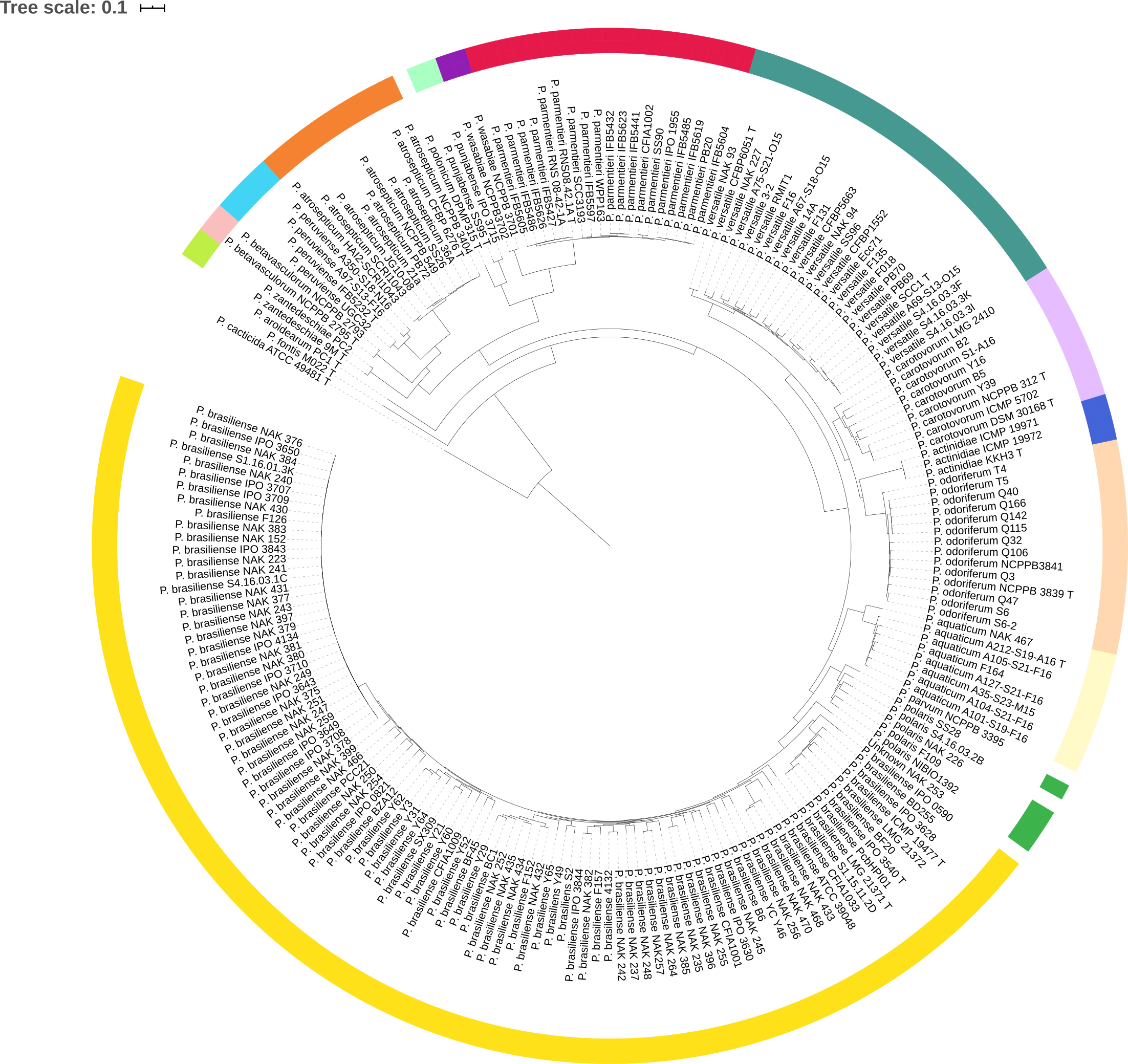Part 3. Phylogeny
Part 3 preparation
If you did not follow part 2 of the tutorial, download the pre-constructed pangenome here.
$ wget http://bioinformatics.nl/pangenomics/tutorial/pecto_dickeya_DB.tar.gz
$ tar xvzf pecto_dickeya_DB.tar.gz
Adding phenotype/metadata to the pangenome
Before we construct the trees, we will add some phenotype data to the pangenome. Once the we have a phylogeny, the information can be included or be used to color parts of the tree. Below is a textfile with data for three phenotypes. The third phenotype, low_temperature, is in this case a made up example! It states whether the strain is capable of growing on (extreme) low temperatures. The phenotype file can be found inside the database directory, add the information to the pangenome by using add_phenotypes.
Genome, species, strain_name, low_temperature
1,P. odoriferum,P. odoriferum Q166, false
2,P. fontis, P. fontis M022, true
3,P. polaris,P. polaris S4.16.03.2B, false
4,P. brasiliense, P. brasiliense S2, true
5,P. brasiliense, P. brasiliense Y49, false
6,D. dadantii, D. dadantii 3937,?
$ pantools add_phenotypes pecto_dickeya_DB pecto_dickeya_DB/phenotypes.txt
Constructing a phylogeny
In this tutorial we will construct three phylogenies, each based on a different type of variation: SNPs, genes and k-mers. Take a look at the phylogeny manuals to get an understanding how the three methods work and how they differ from each other.
Core SNP phylogeny
The core SNP phylogeny will run various Maximum Likelihood models on parsimony informative sites of single-copy orthologous sequences. A site is parsimony-informative when there are at least two types of nucleotides that occur with a minimum frequency of two. The informative sites are automatically identified by aligning the sequences; however, it does not know which sequences are single-copy orthologous. You can identify these conserved sequences by running gene_classification.
$ pantools gene_classification --phenotype=species pecto_dickeya_DB
Open gene_classification_overview.txt and take a look at statistics. As you can see there are 2134 single-copy ortholog groups. Normally, all of these groups are aligned to identify SNPs but for this tutorial we’ll make a selection of only a few groups to accelerate the steps. You can do this in two different ways:
Option 1: Open single_copy_orthologs.csv and remove all node identifiers after the first 20 homology groups and save the file.
$ pantools core_phylogeny --mode=ML pecto_dickeya_DB
Option 2: Open single_copy_orthologs.csv and select the first 20 homology_group node identifiers. Place them in a new file sco_groups.txt and include this file to the function.
$ pantools core_phylogeny --mode=ML -H=sco_groups.txt pecto_dickeya_DB
The sequences of the homology groups are being aligned two consecutive times. After the initial alignment, input sequences are trimmed based on the longest start and end gap of the alignment. The parsimony informative positions are taken from the second alignment and concatenated into a sequence. When opening informative.fasta you can find 6 sequences, the length of the sequences being the number of parsimony-informative sites.
$ iqtree -nt 4 -s pecto_dickeya_DB/alignments/grouping_v1/core_phylogeny/informative.fasta -redo -bb 1000
IQ-tree generates several files, the tree that we later on in the tutorial will continue with is called informative.fasta.treefile. When examining the informative.fasta.iqtree file you can find the best fit model of the data. This file also shows the number of sites that were used, as sites with gaps (which IQ-tree does not allow) were changed into singleton or constant sites.
Gene distance tree
To create a phylogeny based on gene distances (absence/presence), we can simply execute the Rscript that was created by gene_classification.
$ Rscript pecto_dickeya_DB/gene_classification/gene_distance_tree.R
The resulting tree is called gene_distance.tree.
K-mer distance tree
To obtain a k-mer distance phylogeny, the k-mers must first be counted with the kmer_classification function. Afterwards, the tree can be constructed by executing the Rscript.
$ pantools kmer_classification pecto_dickeya_DB
$ Rscript pecto_dickeya_DB/kmer_classification/genome_kmer_distance_tree.R
The resulting tree is written to genome_kmer_distance.tree.
Renaming tree nodes
So far, we used three different types of distances (SNPs, genes, k-mers), and two different methods (ML, NJ) to create three phylogenetic trees. First, lets take a look at the text files. The informative.fasta.treefile only contain genome numbers, bootstrap values and branch lengths but is lacking the metadata. Examining gene_distance.tree file also shows this information but the species names as well, because we included this as a phenotype during gene_classification.
Let’s include the strain identifiers to the core snp tree to make the final figure more informative. Use the rename_phylogeny function to rename the tree nodes.
$ pantools rename_phylogeny --phenotype=strain_name pecto_dickeya_DB pecto_dickeya_DB/alignments/grouping_v1/core_phylogeny/informative.fasta.treefile
Take a look at informative.fasta_RENAMED.treefile, strain identifiers have been added to the tree.
Visualizing the tree in iTOL
Go to https://itol.embl.de and click on “Upload a tree” under the ANNOTATE box. On this page you can paste the tree directly into the tree text: textbox or can click the button to upload the .newick file.
Basic controls iTOL
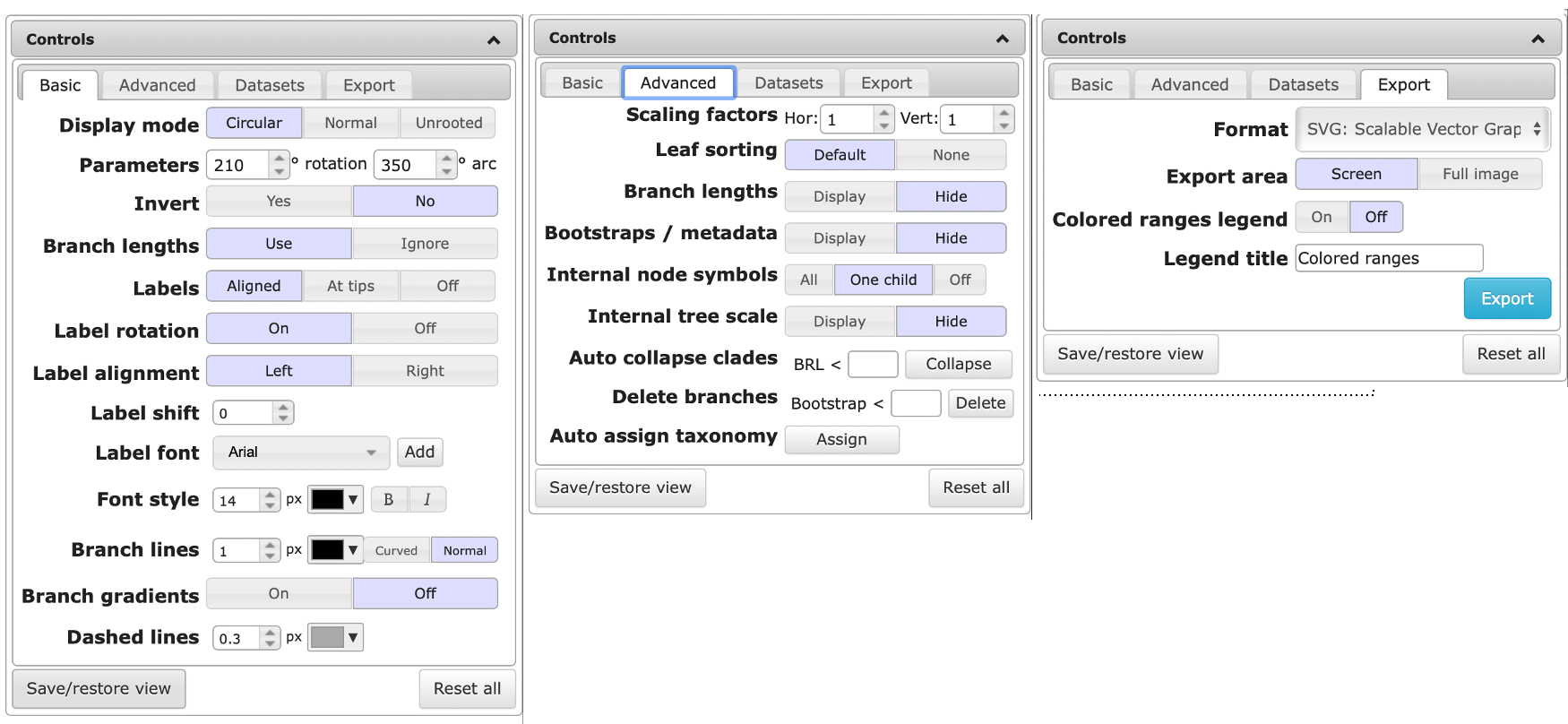
The default way of visualizing a tree is the rectangular view. Depending on the number of genomes, the circular view can be easier to interpret. You can the view by clicking on the “Display Mode” buttons.
Increase the font size and branch width to improve readability
When visualizing a Maximum likelihood (ML) tree, bootstrap values can be displayed by clicking the “Display” button next to Bootstrap/metadata in the Advanced tab of the Control window. This enables you to visualize the values as text or symbol on the branch. or by coloring the branch or adjusting the width.
When you have a known out group or one of the genomes is a clear outlier in the tree, you should reroot the tree. Hover over the name, click it so a pop-up menu appears. Click “tree structure” followed by “Reroot the tree here”.

Clicking on the name of a node in the tree allows you to color the name, branch, or background of that specific node.
When you’re happy the way your tree looks, go to the Export tab of the Control window. Select the desired output format, click on the “Full image” button and export the file to a figure.
Refresh the webpage to go back to the default view of your tree.
Create iTOL templates
In iTOL it is possible to add colors to the tree by coloring the
terminal nodes or adding an outer ring. The PanTools function
create_tree_template is able
to create templates that allows for easy coloring (with maximum of 20
possible colors). If the function is run without any additional
argument, templates are created for trees that only contain genome
numbers (e.g. k-mer distance tree). Here we want to color the (renamed)
core SNP tree with the ‘low_temperature’ phenotype. Therefore, the
--phenotype strain_name must be included to the function.
$ pantools create_tree_template pecto_dickeya_DB # Run this command when the tree contains genome numbers only
$ pantools create_tree_template --phenotype=strain_name pecto_dickeya_DB
Copy the two low_temperature.txt files from the label/strain_name/ and ring/strain_name/ directories to your personal computer. Click and move the ring template file into the tree visualization webpage.
The resulting tree should look this when: the tree is rooted with the Dickeya genome, bootstrap values are displayed as text and the ring color template was included.

Tree coloring is especially useful for large datasets. An example is shown in the figure below, where members of the same species share a color.