Part 4. Characterization
Part 4 preparation
PanTools v3 is required to follow this part of the tutorial. In addition, MAFFT and R (and a few packages) need to be installed and set to your $PATH. Everything should already be correctly installed if you use the conda environment. Validate if the tools are executable by using the following commands.
$ Rscript --help
$ mafft -h
We assume a PanTools alias was set during the
installation. This allows
PanTools to be executed with pantools rather than the full path to the jar
file. If you don’t have an alias, either set one or replace the pantools
command with the full path to the .jar file in the tutorials.
Input data
Genome |
Name |
Accession |
Length |
Sequences |
Genes |
|---|---|---|---|---|---|
1 |
|
5.09 Mb |
66 |
4510 |
|
2 |
|
4.15 Mb |
107 |
3723 |
|
3 |
|
4.86 Mb |
65 |
4442 |
|
4 |
|
4.84 Mb |
37 |
4367 |
|
5 |
|
4.70 Mb |
31 |
4231 |
|
6 |
|
4.92 Mb |
1 |
4281 |
To demonstrate how to use the PanTools functionalities we use a small dataset of six bacteria to avoid long runtimes. Download a pre-constructed pangenome or test your new skills and construct a pangenome yourself using the fasta and gff files.
Option 1: Download separate genome and annotation files
$ wget http://bioinformatics.nl/pangenomics/tutorial/pecto_dickeya_input.tar.gz
$ tar -xvzf pecto_dickeya_input.tar.gz
$ gzip -d pecto_dickeya_input/annotations/*
$ gzip -d pecto_dickeya_input/genomes/*
$ gzip -d pecto_dickeya_input/functions/*
$ pantools build_pangenome pecto_dickeya_DB pecto_dickeya_input/genomes.txt
$ pantools add_annotations --connect pecto_dickeya_DB pecto_dickeya_input/annotations.txt
$ pantools group --relaxation=4 pecto_dickeya_DB
Option 2: Download the pre-constructed pangenome
$ wget http://bioinformatics.nl/pangenomics/tutorial/pecto_dickeya_DB.tar.gz
$ tar -xvzf pecto_dickeya_DB.tar.gz
Adding phenotype/metadata to the pangenome
Before starting with the analysis, we will add some phenotype data to the pangenome. Phenotypes allow you to find similarities for a group of genomes sharing a phenotype as well as identifying variation between different phenotypes. Below is a textfile with data for three phenotypes. The third phenotype, low_temperature, is in this case a made up example! It states whether the strain is capable of growing on (extreme) low temperatures. The phenotype file can be found inside the database directory or create a new file using the text from the box below. Add the phenotype information to the pangenome using add_phenotypes.
Genome, species, strain_name, low_temperature
1,P. odoriferum,P. odoriferum Q166, false
2,P. fontis, P. fontis M022, true
3,P. polaris,P. polaris S4.16.03.2B, false
4,P. brasiliense, P. brasiliense S2, true
5,P. brasiliense, P. brasiliense Y49, false
6,D. dadantii, D. dadantii 3937,?
$ pantools add_phenotypes pecto_dickeya_DB pecto_dickeya_input/phenotypes.txt
Metrics and general statistics
After building or uncompressing the pangenome, run the metrics functionality to produce various statistics that should verify an errorless construction.
$ pantools metrics pecto_dickeya_DB
Open metrics_per_genome.csv with a spreadsheet tool (Excel, Libreoffice, Google sheets) and make sure the columns are split on commas. You may easily notice the many empty columns in this table as these type of annotations or features are not included in the database (yet). Functional annotations are incorporated later in this tutorial. Columns for features like exon and intron will remain empty as bacterial coding sequences are not interrupted.
Gene classification
With the gene_classification functionality you are able to organize the gene repertoire into the core, accessory or unique part of the pangenome.
Core, a gene is present in all genomes
Unique, a gene is present in a single genome
Accessory, a gene is present in some but not all genomes
$ pantools gene_classification pecto_dickeya_DB
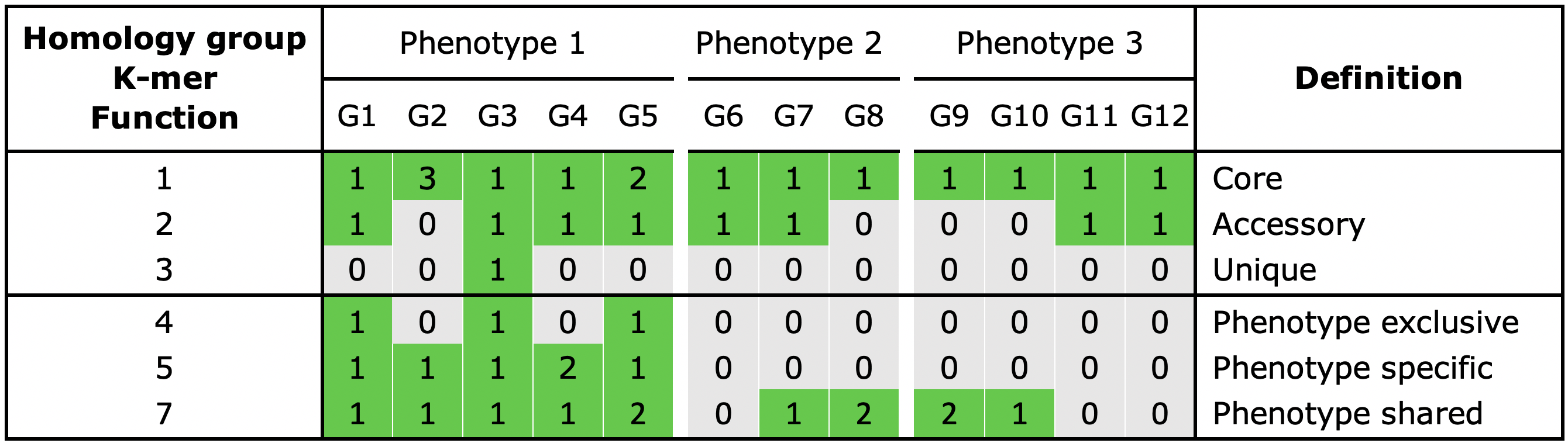
Take a look in gene_classification_overview.txt. Here you can find the number of classified homology groups and genes on a pangenome level but also for individual genomes.
Open additional_copies.csv with a spreadsheet tool. This file can be useful to identify duplicated genes in relation to other genomes.
The default criteria to call a group core is presence in all genomes
where unique is only allowed to be present in one genome. These two
categories are highly influenced by annotation quality, especially in
large pangenomes. Luckily, the threshold for core and unique groups can
easily be adjusted. Let’s consider genes to be core when present in only
five of the six genomes by setting the --core-threshold argument.
$ pantools gene_classification --core-threshold=85 pecto_dickeya_DB
Look in gene_classification_overview.txt again to observe the increase of core groups/genes at the cost of accessory groups.
For this pangenome, the Dickeya genome is considered an outgroup to
the five Pectobacterium genomes. While this outgroup is needed to root
and analyze phylogenetic trees (tutorial part 5), it affects the number
classified groups for the all other genomes. Use --include or
--exclude to exclude the Dickeya genome.
$ pantools gene_classification --include=1-5 pecto_dickeya_DB
$ pantools gene_classification --exclude=6 pecto_dickeya_DB
Take a look in gene_classification_overview.txt one more time to examine the effect of excluding this genome. The total number of groups in the analysis is lower now but the number of core and unique genes have increased for the five remaining genomes.
When phenotype information is used in the analysis, three additional categories can be assigned to a group:
Shared, a gene present in all genomes of a phenotype
Exclusive, a gene is only present in a certain phenotype
Specific, a gene present in all genomes of a phenotype and is also exclusive
Include a --phenotype argument to find genes that are exclusive for
a certain species.
$ pantools gene_classification --phenotype=species pecto_dickeya_DB
Open gene_classification_phenotype_overview.txt to see the number of classified groups for the species phenotype.
Open phenotype_disrupted.csv in a spreadsheet tool. This file explains exactly why a homology groups is labeled as phenotype shared and not specific.
Open phenotype_additional_copies.csv in a spreadsheet tool. Similarly to phenotype_additional.csv this file shows groups where all genomes of a certain phenotype have additional gene copies to (at least one of) the other phenotypes.
Each time you run the gene_classification function, multiple files are created that contain node identifiers of a certain homology group category. These files can be given to other PanTools functions for a downstream analysis, for example, sequence alignment, phylogeny, or GO enrichment. We will use one of the files later in this tutorial.
Pangenome structure
With the previous functionality we identified the core, accessory and unique parts of the pangenome. Now we will use the pangenome_size_genes function to observe how these numbers are reached by simulating the growth of the pangenome. Simulating the growth helps explaining if a pangenome should be considered open or closed. An pangenome is called open as long as a significant number of new (unique) genes are added to the total gene repertoire. The openness of a pangenome is usually tested using Heap’s law. Heaps’ law (a power law) can be fitted to the number of new genes observed when increasing the pangenome by one random genome. The formula for the power law model is n = k x N-a, where n is the newly discovered genes, N is the total number of genomes, and k and a are the fitting parameters. A pangenome can be considered open when a < 1 and closed if a > 1.
The outcome of the function can again be controlled through command line
arguments. Genomes can be excluded from the analysis with --exclude.
You can set the number of iterations with --loops.
$ pantools pangenome_structure pecto_dickeya_DB
The current test set of six bacterial genomes is not representative of a full-sized pangenome. Therefore we prepared the results for the structure simulation on a set of 197 Pectobacterium genomes. The runtime of the analysis using 10.000 loops and 24 threads was 1 minute and 54 seconds. Download the files here, unpack the archive and take a look at the files.
$ wget wget http://bioinformatics.nl/pangenomics/tutorial/pectobacterium_structure.tar.gz
$ tar -xvf pectobacterium_structure.tar.gz
Normally you still have to run the R scripts to create the output figures and determine the openness of the pangenome.
cd pectobacterium_structure
$ Rscript pangenome_growth.R
$ Rscript gains_losses_median_and_average.R
$ Rscript heaps_law.R
Take a look at the plot. In core_accessory_unique_size.png, the number of classified groups are plotted for any of the genome combination that occurred during the simulation. For the core_accessory_size.png plots, the number of unique groups is combined with accessory groups.
The gains_losses.png files display the average and mean group gain and loss between different pangenome sizes. The line of the core starts below zero, meaning for every random genome added, the core genome decreases by a number of X genes.
The alpha value of heaps' law is written to heaps_law_alpha.txt. Click here to reveal the result.
With an alpha of 0.53 the Pectobacterium pangenome has an
open structure, which is typical for many bacterial species due
their extensive exchange of genetic information. Performing this
analysis on a set of closely related animal or plant genomes usually
results in a closed pangenome, indicating a limited gene pool.
Functional annotations
PanTools is able to incorporate functional annotations into the pangenome by reading output of various functional annotation tools. In this tutorial we only include annotations from InterProScan. Please see the add_functions manual to check which other tools are available. To include the annotations, create a file functions.txt using text from the box below and add it to the command line argument.
1 YOUR_PATH/GCF_002904195.1.gff3
2 YOUR_PATH/GCF_000803215.1.gff3
3 YOUR_PATH/GCF_003595035.1.gff3
4 YOUR_PATH/GCF_000808375.1.gff3
5 YOUR_PATH/GCF_000808115.1.gff3
6 YOUR_PATH/GCA_000147055.1.gff3
$ pantools add_functions pecto_dickeya_DB functions.txt
PanTools will ask you to download the InterPro database. Follow the steps and execute the program again.
The complete GO, PFAM, Interpro and TIGRFAM, databases are now integrated in the graph database after. Genes with a predicted function have gained a relationship to that function node. Retrieving a set of genes that share a function is now possible through a simple cypher query. If you would run metrics again, statistics for these type functional annotations are calculated. To create a summary table for each type of functional annotation, run function_overview.
$ pantools function_overview pecto_dickeya_DB
In function_overview_per_group.csv you can navigate to a homology group or gene to see the connected functions. You can also search in the opposite direction, use one of the created overview files for a type of functional annotation and quickly navigate to a function of interest to find which genes are connected.
GO enrichment
We go back to the output files from gene classification that only
contain node identifiers. We can retrieve group functions by providing
one the files to
group_info with the
--homology-groups argument. However, interpreting groups by
assessing each one individually is not very practical. A common
approach to discover interesting genes from a large set is
GO-enrichment. This statistical method enables the identification of
genes sharing a function that are significantly over or
under-represented in a given gene set compared to the rest of the
genome. Let’s perform a GO enrichment
on homology groups of the core genome.
Phenotype: P._brasiliense, 2 genomes, threshold of 2 genomes
1278537,1282642,1283856,1283861,1283862,1283869,1283906,1283921,1283934,1283941,1283945,1283946
$ pantools group_info pecto_dickeya_DB brasiliense_groups.csv
$ pantools go_enrichment pecto_dickeya_DB brasiliense_groups.csv
Open go_enrichment.csv with a spreadsheet tool. This file holds GO terms found in at least one of the genomes, the p-value of the statistical test and whether it is actually enriched after the multiple testing correction. as this is different for each genome a function might enriched in one genome but not in another.
A directory with seperate output files is created for each genome, open go_enrichment.csv for the genome 4 or 5 in a spreedsheet. Also take a look at the PDF files that visualize part of the Gene ontology hierarchy.
Classifying functional annotations
Similarly to classifying gene content, functional annotations can be categorized using functional_classification. This tool provides an easy way to identify functions shared by a group of genomes of a certain phenotype but can also be used to identify core or unique functions. The functionality uses the same set of arguments as gene_classification. You can go through the same steps again to see the effect of changing the arguments.
$ pantools functional_classification pecto_dickeya_DB
$ pantools functional_classification --core-threshold=85 pecto_dickeya_DB
$ pantools functional_classification --exclude=6 pecto_dickeya_DB
$ pantools functional_classification --phenotype=species pecto_dickeya_DB
Sequence alignment
In the final part of this tutorial we will test the alignment function by aligning homology groups. PanTools is able to align genomic regions, genes and proteins to identify SNPs or amino acid changes with msa.
Start with the alignment of protein sequences from the 12 P. brasiliense specific homology groups.
$ pantools msa --mode=protein --method=per-group -H=brasiliense_groups.csv pecto_dickeya_DB
Go to the pecto_dickeya_DB/alignments/grouping_v1/groups/ directory and select one of homology groups and check if you can find the following files
The alignments are written to prot_trimmed.fasta and prot_trimmed.afa.
A gene tree is written to prot_trimmed.newick
prot_trimmed_variable_positions.csv located in the var_inf_positions subdirectory. This matrix holds every variable position of the alignment; the rows are the position in the alignment and the columns are the 20 amino acids and gaps.
The identity and similarity (for proteins) is calculated between sequences and written to tables in the similarity_identity subdirectory.
Run the function again but include the --no-trimming argument.
$ pantools msa --no-trimming --mode=protein --method=per-group -H=brasiliense_groups.csv pecto_dickeya_DB
The output files are generated right after the first alignment without trimming the sequences first. The file names differ from the trimmed alignments by the ’_trimmed’ substring.
Run the function again but exclude the --mode=protein and
--no-trimming arguments. When no additional arguments are included
to the command, both nucleotide and protein sequences are aligned two
consecutive times.
$ pantools msa --method=per-group -H=brasiliense_groups.csv pecto_dickeya_DB
Again, the same type of files are generated but the output files from nucleotide sequence can be recognized by the ‘nuc_’ substrings. The matrix in nuc_trimmed_variable_positions.csv now only has columns for the four nucleotides and gaps.
Finally, run the function one more time but include a phenotype. This allows you to identify phenotype specific SNPs or amino acid changes.
$ pantools msa --no-trimming --phenotype=low_temperature -H=brasiliense_groups.csv pecto_dickeya_DB
Open the nuc- or prot_trimmed_phenotype_specific_changes.info file inside one of the homology group output directories.
Besides the functionalities in this tutorial, PanTools has more useful functions that may aid you in retrieving more specific information from the pangenome.
Identify shared k-mers between genomes with kmer_classification.
Find co-localized genes in a set of homology groups: locate_genes.
Mapping short reads against the pangenome with map.
In part 5 of the tutorial we explore some of the phylogenetic methods implemented in PanTools.