Phylogeny
There are six different methods implemented which can create phylogenetic trees. The consensus tree method creates a Maximum per-Locus Quartet-score Species Tree (MLQST) from a set of gene trees. The other five methods use a Neighbour-joining (NJ) or Maximum Likelihood (ML) algorithm to infer the phylogeny.
Core phylogeny (ML)
K-mer distance tree (NJ)
Gene distance tree (NJ)
ANI (NJ)
MLSA (ML)
All functions produce tree files in Newick format that can be visualized with iTOL or any other phylogenetic tree visualization software.
Core phylogeny
Infer a Maximum likelihood (ML) or Neighbour-Joining (NJ) phylogeny from SNPs identified from single copy orthologous genes. This function requires single-copy homology groups which are automatically detected if gene_classification was run before. The homology groups are aligned in two consecutive rounds with msa.
When using --clustering-mode ML, parsimony informative positions
are extracted from the trimmed alignments and concatenated into single
continuous sequence per genome. IQ-tree infers the ML tree with minimum
of 1000 bootstrap iterations.
The --clustering-mode NJ method counts the total and shared number
of variable sites between two genomes in the alignment and calculates a
Jaccard distance (0-1):
Required software
Please cite the appropriate tool(s) when using the core phylogeny in your research.
Parameters
<databaseDirectory> |
Path to the database root directory. |
Options
|
Number of parallel working threads, default is the number of cores or 8, whichever is lower. |
|
Only include a selection of genomes. |
|
Exclude a selection of genomes. |
|
A file with homology group node identifiers of single copy groups.
Default is single_copy_orthologs.csv, generated in the previous
gene_classification run.
(Mutually exclusive with |
|
A comma separated list of homology group node identifiers of single
copy groups. Default is single_copy_orthologs.csv, generated in
the previous gene_classification run.
(Mutually exclusive with |
|
Use proteins instead of nucleotide sequences. |
|
Include phenotype information in the resulting phylogeny. |
|
Maximum likelihood (–mode ML) or Neighbour joining (–mode NJ). Default is ML. |
Example commands
$ pantools core_phylogeny -t=24 tomato_DB
$ pantools core_phylogeny -t=24 --clustering-mode=NJ --protein tomato_DB
$ pantools core_phylogeny -t=24 -m=ML -p=resistance tomato_DB
Output
Output files are written to the core_snp_tree directory in the database.
sites_per_group.csv, number of parsimony informative and variable sites per homology group.
When --clustering-mode NJ is included
core_snp_NJ_tree.R, Rscript to create NJ tree from distances based on shared sites. Two distances can be selected, based on variable sites and parsimony informative sites.
shared_informative_positions.csv, table with total number of shared parsimony informative sites between genomes.
shared_variable_positions.csv, table with total number of shared variable sites between genomes.
When --clustering-mode ML is included
informative.fasta, nucleotides from parsimony informative sites of the alignments, concatenated into a single sequences per genomes.
variable.fasta, nucleotides from variable sites of the alignment, concatenated into a single sequences per genomes.
A command is generated which can be used to execute IQ-tree and infer the phylogeny on informative.fasta.
informative.fasta.iqtree, IQ-tree log file.
informative.fasta.treefile, the ML phylogeny.
informative.fasta.splits.nex, the splits graph. With ideal data, this file is a tree, whereas data with conflicting phylogenetic signals will result in a tree-like network. This type of tree/network can be visualized with a tool like SplitsTree
K-mer distance tree
A NJ phylogeny of k-mer distances can be created by executing the Rscript generated by k-mer_classification.
Three types of distances can be selected to infer the phylogeny. The first two distances are Jaccard distances (0-1): one considering only distinct k-mers and the other using all k-mers. The distance from distinct k-mers ignores additional copies of a k-mer.
We observed an exponential increase in the k-mer distance as the evolutionary distance between two genomes increases. So in the case of more distant genomes, the depicted clades are still correct but the extreme long branch lengths make the tree hard to decipher. To normalize the numbers, we implemented the MASH distance. Distance = −1/𝑘 * ln(J), where k is the k-mer length; J is the jaccard index (of distinct k-mers).
$ Rscript genome_kmer_distance_tree.R
Output file
The phylogenetic tree genome_kmer_distance_tree.tree is written to the kmer_classification directory in the database.
Consensus tree
Create a consensus tree by combining gene trees from homology groups using ASTRAL-Pro. Gene trees are created from all sequences in an homology groups, no genomes can be skipped.
Required software
Please cite MAFFT, FastTree and ASTRAL-Pro when using the consensus tree in your research.
Parameters
<databaseDirectory> |
Path to the database root directory. |
Options
|
Number of parallel working threads, default is the number of cores or 8, whichever is lower. |
|
A file with homology group node identifiers. Default is all
homology groups. (Mutually exclusive with |
|
A comma separated list of homology group node identifiers. Default
is all homology groups. (Mutually exclusive with
|
|
Allow polytomies for ASTRAL-PRO. |
Example commands
$ pantools consensus_tree -t=24 apple_DB
$ pantools consensus_tree -H=apple_DB/gene_classification/group_identifiers/all_homology_groups.csv apple_DB
$ pantools consensus_tree -H=apple_DB/gene_classification/group_identifiers/core_homology_groups.csv apple_DB
$ pantools consensus_tree -H=apple_DB/gene_classification/group_identifiers/accessory_homology_groups.csv apple_DB
Output
Output files are written to the consensus_tree directory in the database.
all_trees.hmgroups.newick, all gene trees of homology groups included in the analysis, combined into a single file.
consensus_tree.astral-pro.newick, the output consensus tree from ASTRAL-Pro.
Relevant literature
Gene distance tree
A NJ phylogeny of gene distances is created by executing the Rscript generated by gene_classification.
Shared genes between genomes are identified through homology groups. Two Jaccard distance (0-1) can be used to infer a tree: one considering only distinct genes and the other using all genes. The distance from distinct genes ignores additional gene copies in an homology group.
$ Rscript gene_distance_tree.R
Output file
The phylogenetic tree gene_distance_tree.tree is written to the gene_classification directory in the database.
ANI
Average Nucleotide Identity (ANI) is a measure of nucleotide-level genomic similarity between the coding regions of two prokaryotic genomes. Two very fast ANI estimation tools (fastANI and MASH) are implemented and are able to perform the pairwise comparisons between genomes in the pangenome. To convert the ANI score into a distance (0-1), the scores are transformed by \(1-(ANI/100)\).
Required software
The required software depends on the tool you want to use. Please cite the appropriate tool when using the ANI tree in your research.
Parameters
<databaseDirectory> |
Path to the database root directory. |
Options
|
Number of parallel working threads, default is the number of cores or 8, whichever is lower. MASH is always single threaded (and currently not parallelized yet). |
|
Only include a selection of genomes. |
|
Exclude a selection of genomes. |
|
Software to calculate ANI score (default: MASH). |
|
Include phenotype information in the phylogeny. |
Example commands
$ pantools ani pecto_DB
$ pantools ani --phenotype=species_name --mode=fastani pecto_DB
$ pantools ani --exclude=4,5,6 --mode=mash pecto_DB
Output
Output files are written to the ANI directory in the database.
ANI_scores.csv, a table with ANI scores for all genome pairs.
ANI_distance_matrix.csv, a table with the ANI distances (1-ANI). This matrix is read by ANI_tree.R.
ANI_tree.R, Rscript to generate NJ tree from ANI distances
Find closest typestrain
Compares bacterial strains to the typestrain when this information is available in a pangenome database.
Add the ‘typestrain’ phenotype to the pangenome with add_phenotypes. You only have to include typestrains names, other genomes can be left empty as shown in the example below, five genomes with three different typestrains.
Run the ANI function
The ‘typestrain’ phenotype is recognized, and typestrain_comparison.csv is created. This file contains the highest score of each genome(5) against all the included typestrains and states whether the score is above 95%.
Genome,typestrain
1,Salmonella choleraesuis NCTC 5735
2,Salmonella enteritidisi NCTC 12694
3,
4,Salmonella paratyphi NCTC 5702
5,
Relevant literature
MLSA
Within PanTools you can perform a Multilocus sequence analysis (MLSA) by running three consecutive functions:
Step 1 Search for genes
Find your genes of interest in the pangenome and extract their
nucleotide and protein sequence. A regular search is not case sensitive
but the gene names must exactly match the given input name. For example,
searching a gene with ‘sonic1’ as query will not be able find ‘sonic’,
but is able to find Sonic1, SONIC1 or sOnIc1. Including the
--extensive argument allows a more relaxed search and using
‘sonic’ will now also find gene name variations as ‘sonic1’, ‘sonic3’
etc.. For this function it is important that genomes are annotated by a
method that follow the rules for genetic nomenclature, so there are no
differences in the naming of genes.
To gain insight in which genes are appropriate for this analysis, run
gene_classification with the
--mlsa argument. This method creates a list of genes that have
same gene name, are present in all (selected) genomes and are placed in
the single-copy homology group. Using genes from this list guarantees a
successful MLSA.
Possible generated warnings during gene search
When a gene is included that is not on the list of suitable genes, it is not necessarily unusable but possibly requires manual . This function generates a log file with the issues and explains the user what to do.
Gene is not found in every genome. Consider using
--extensive. The gene is not suitable with the current genome selection when this argument was already included.The found genes are placed in different homology groups. A directory named the gene name is created where sequences are stored in a separate file per homology group. When one of the groups is single copy orthologous, it is automatically selected. With multiple correct single-copy groups, the first is selected. If no single-copy groups are found, this gene is probably not a suitable candidate based on the high divergence. If you are determined to use the gene, align and infer a gene tree on all_sequences.fasta to identify appropriate sequences.
At least one gene has an additional copy. The extra copies must be removed from the output file if you want to include this gene in the analysis. Find the copies that stand out by aligning and inferring a gene tree of the homology group.
Parameters
<databaseDirectory> |
Path to the database root directory. |
Options
|
One or multiple gene names, separated by a comma. |
|
Only include a selection of genomes. |
|
Exclude a selection of genomes. |
|
Perform a more extensive gene search. |
Example commands
$ pantools mlsa_find_genes -g=dnaX,gapA,recA bacteria_DB
$ pantools mlsa_find_genes --extensive --genes=gapA bacteria_DB
Output
Output files are written to the mlsa/input/ directory in the database. For each gene name that was included, a nucleotide and protein and FASTA file is created that holding the sequences found in all genomes.
mlsa_find_genes.log, when one or multiple warnings are given they are placed in this log file. File is not created when there aren’t any warnings.
Step 2 Concatenate genes
Concatenate sequences obtained by
mlsa_find_genes into a single
sequence per genome. The --genes argument is required, but the
selection of gene names is allowed to be a sub-selection of the earlier
selection.
Proteins are aligned with MAFFT
The longest gap at the start and end of each protein alignment is identified.
Nucleotide sequences are trimmed accordingly
Trimmed nucleotide sequence are concatenated into a single sequence per genome.
Required software
Parameters
<databaseDirectory> |
Path to the database root directory. |
Options
|
One or multiple gene names, separated by a comma. |
|
Number of threads for MAFFT, default is the number of cores or 8, whichever is lower. |
|
Only include a selection of genomes. |
|
Exclude a selection of genomes. |
|
Name of the phenotype. |
Example commands
$ pantools mlsa_concatenate --genes=dnaX,gapA bacteria_DB
$ pantools mlsa_concatenate --g=dnaX,gapA,recA --e=1,2,10-25 bacteria_DB
Output
The output file is stored in /database_directory/mlsa/input/
concatenated.fasta, file holding one concatenated sequence per genome.
Step 3 Run MLSA
Run MAFFT and IQ-tree on the concatenated nucleotide sequences from mlsa_concatenate to create an unrooted ML tree with 1,000 bootstrappings.
Required software
Please cite the MAFFT and IQ-tree when using the MLSA in your research.
Parameters
<databaseDirectory> |
Path to the database root directory. |
Options
|
Number of threads for MAFFT and IQ-tree, default is the number of cores or 8, whichever is lower. |
|
Only include a selection of genomes. |
|
Exclude a selection of genomes. |
|
Add phenotype information/values to the phylogeny. Allows the identification of phenotype specific SNPs in the alignment. |
Example commands
$ pantools mlsa bacteria_DB
$ pantools mlsa -t=24 -p=species bacteria_DB
Output
Input and output files are written to the mlsa/output/ directory in the database.
mlsa.afa, the alignment in CLUSTAL format.
mlsa.fasta, the alignment in FASTA format.
mlsa.fasta.treefile, the (ML) phylogeny created by IQ-tree in Newick format.
When a --phenotype is included
nuc_phenotype_specific_changes.info, the positions of phenotype specific substitutions in the alignment.
The var_inf_positions directory holds files related to the counting variable positions of the alignment.
nuc_variable_positions.csv, a table with the counts of A, T, C, G, or gap for every variable position in the alignment
informative_nuc_distance.csv, a table with distances calculated from parsimony informative positions in the alignment.
informative_nuc_site_counts.csv, a table with number of shared parsimony informative positions between genomes.
variable_nuc_distance.csv, a table with distances calculated from variable positions in the alignment.
variable_nuc_site_counts.csv, a table with number of shared positions between genomes.
Edit Phylogeny
Rename phylogeny
Update or the terminal nodes (leaves) of a phylogenic tree. This is
useful when you already constructed a tree but forgot to include a
phenotype or to update the tree with a different phenotype. When no
--phenotype is included, the node values are changed to genome
numbers.
Parameters
<databaseDirectory> |
Path to the database root directory. |
<treeFile> |
A phylogenetic tree in newick or nexus format. The tree must be generated by PanTools. |
Options
|
Exclude genome numbers from the terminal nodes (leaves). |
|
The phenotype used to rename the terminal nodes (leaves) of the selected tree. |
Example commands
$ pantools rename_phylogeny bacteria_DB core_snp.tree
$ pantools rename_phylogeny --phenotype=species bacteria_DB bacteria_DB/ANI/fastANI/ani.tree
Output file
A new phylogenetic tree is written to the directory of the selected input tree:
When the original file is called ‘old_tree.newick’, a new tree is created with filename ‘old_tree_RENAMED.newick’.
Root phylogeny
All phylogenetic trees that come from the PanTools functionalities are
unrooted. This function is able to create a new rooted tree simply by
selecting one of the external (terminal) nodes via --node. The
included number or string should match exactly one node in the phylogeny
or the program will not execute.
Required software
Parameters
<databaseDirectory> |
Path to the database root directory. |
<treeFile> |
A phylogenetic tree in newick format. The tree must be generated by PanTools. |
Options
|
The name of the terminal node that will root the tree. |
Example commands
$ pantools root_phylogeny --node=1 bacteria_DB core_snp.tree
$ pantools root_phylogeny --node=1_A.thaliana bacteria_DB core_snp.tree
$ pantools root_phylogeny -n=1_1 bacteria_DB kmer.tree
$ Rscript reroot.R
Output file
A new phylogenetic tree is written to the same location as the provided input file
When the original tree is called ‘tree.newick’, the new file is named ‘tree_REROOTED.newick’.
Create tree template
Creates ‘ring’ and ‘colored range’ ITOL templates based on phenotypes for the visualization of phylogenies in iTOL. Phenotypes must already be included in the pangenome with the add_phenotypes functionality. How to use the template files in iTOL can be found in one of the tutorials.
If you run this function without a --phenotype argument, templates
are created for trees that contain only genome numbers as node labels.
When there is a --phenotype included, templates are created where
the leaves are named according to the selected phenotype but are
coloured by one of the other phenotypes in the pangenome. For example,
you originally used the ‘species name’ as a phenotype to construct the
phylogeny but want them to be coloured by the ‘pathogenicity’ phenotype.
More information about ITOL templates can be found on their own webpage.
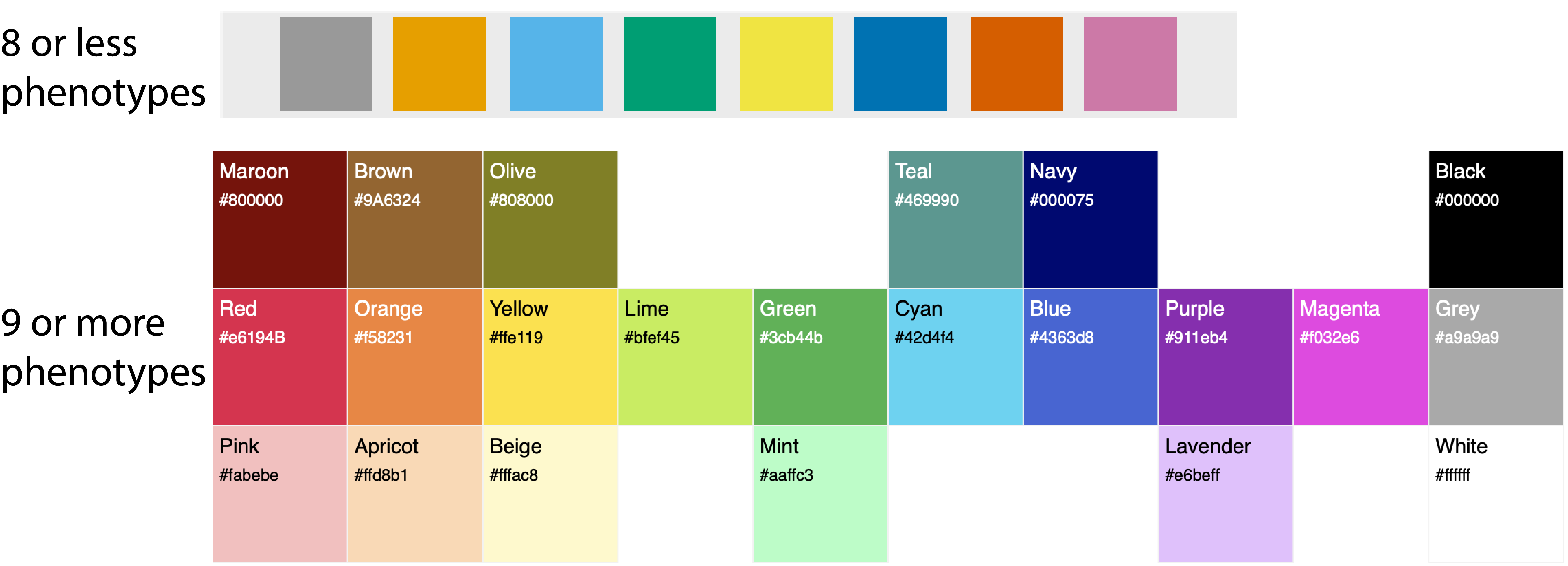
There is a maximum of 20 possible colors that are used in the following order:
Color (> 8 phenotypes) |
Hexadecimal color |
Color (≤ 8 phenotypes) |
Hexadecimal color |
|
|---|---|---|---|---|
1 |
Pink |
#fabebe |
Orange |
#E69F00 |
2 |
Lime |
#bfef45 |
Sky blue |
#56B4E9 |
3 |
Cyan |
#42d4f4 |
Bluish green |
#009E73 |
4 |
Apricot |
#ffd8b1 |
Yellow |
#F0E442 |
5 |
Mint |
#aaffc3 |
Blue |
#0072B2 |
6 |
Beige |
#fffac8 |
Vermilion |
#D55E00 |
7 |
Lavender |
#e6beff |
Reddish purple |
#CC79A7 |
8 |
Teal |
#469990 |
Grey |
#999999 |
9 |
Red |
#e6194B |
||
10 |
Orange |
#f58231 |
||
11 |
Yellow |
#ffe119 |
||
12 |
Green |
#3cb44b |
||
13 |
Blue |
#4363d8 |
||
14 |
Purple |
#911eb4 |
||
15 |
Grey |
#a9a9a9 |
||
16 |
Maroon |
#800000 |
||
17 |
Olive |
#808000 |
||
18 |
Brown |
#9A6324 |
||
19 |
Navy |
#000075 |
||
20 |
Magenta |
#f032e6 |
Figures were copied from:
Parameters
<databaseDirectory> |
Path to the database root directory. |
Options
|
Assign a color to a phenotypes with a minimum amount of genomes (default: 2). |
|
Use the names from this phenotype. |
Example commands
$ pantools create_tree_template bacteria_DB
$ pantools create_tree_template --phenotype=flowering --color=1 bacteria_DB
$ pantools create_tree_template --phenotype=root_morph --color=3 bacteria_DB
Output
Output files are written to the create_tree_template directory in the database.
When no phenotype information is included, a directory ‘genome_numbers’ is created where the templates are stored.
When a
--phenotypeis included, a directory (named after the phenotype) is created where the templates are stored.
The template files are named after the phenotypes, therefore the colors are based on that phenotype as well.