Construct pangenome
Build pangenome
Build a pangenome out of a set of genomes.
Required software
Required arguments
--database-path/-dp Path to the pangenome database.--genomes-file/-gf A text file containing paths to FASTA files
of genomes to be added to the pangenome; each on a separate line.Optional arguments
--kmer-size/-ks Size of k-mers, allowed to be 6 <= K_SIZE <=
255. By not giving this argument, the most optimal k-mer size is
calculated automatically.
Example input file
/always/genome1.fasta
/use_the/genome2.fasta
/full_path/genome3.fasta
Example command
$ pantools build_pangenome -dp tomato_DB -gf tomato_3.txt
Relevant literature
Add annotations
Construct or expand the annotation layer of an existing pangenome. The layer consists of genomic features like genes, mRNAs, proteins, tRNAs etc. PanTools is only able to read General Feature Format (GFF) files.
Multiple annotations can be assigned to a single genome; however, only
one annotation a time can be included in an analysis. The most recently
included annotation of a genome is included as default, unless a
different annotation is specified via --annotations-file, see the
explanation
below
Required arguments
--database-path/-dp Path to the pangenome database.--annotations-file/-af A text file with on each line a genome
number and the full path to the corresponding annotation file,
separated by a space.Optional arguments
--connect-annotations/-ca Connect the annotated genomic features
to nucleotide nodes in the DBG.
Example command
$ pantools add_annotations -dp tomato_DB -af annotations.txt
Output
The annotated features are incorporated in the graph. Output files are written to the database directory.
annotation_overview.txt, a summary of the GFF files incorporated in the pangenome
annotation.log, a list of misannotated feature identifiers.
Example input file
Each line of the file starts with the genome number followed by the full path to the annotation file. The genome numbers match the line number of the file that you used to construct the pangenome.
1 /always/genome1.gff
2 /use_the/genome2.gff
3 /full_path/genome3.gff
CP004621.1 Genbank gene 44836 45753 . - . ID=gene99;Name=RPL23A;end_range=45753,.;gbkey=Gene;gene=RPL23A;gene_biotype=protein_coding;locus_tag=H754_YJM320B00023;partial=true;start_range=.,44836
CP004621.1 Genbank mRNA 44836 45753 . - . ID=rna99;Parent=gene99;gbkey=mRNA;gene=RPL23A;product=Rpl23ap
CP004621.1 Genbank exon 45712 45753 . - . ID=id112;Parent=rna99;gbkey=mRNA;gene=RPL23A;product=Rpl23ap
CP004621.1 Genbank exon 44836 45207 . - . ID=id113;Parent=rna99;gbkey=mRNA;gene=RPL23A;product=Rpl23ap
CP004621.1 Genbank CDS 45712 45753 . - 0 ID=cds92;Parent=rna99;Dbxref=SGD:S000000183,NCBI_GP:AJQ01854.1;Name=AJQ01854.1;Note=corresponds to s288c YBL087C;gbkey=CDS;gene=RPL23A;product=Rpl23ap;protein_id=AJQ01854.1
CP004621.1 Genbank CDS 44836 45207 . - 0 ID=cds92;Parent=rna99;Dbxref=SGD:S000000183,NCBI_GP:AJQ01854.1;Name=AJQ01854.1;Note=corresponds to s288c YBL087C;gbkey=CDS;gene=RPL23A;product=Rpl23ap;protein_id=AJQ01854.1
Select specific annotations for analysis
Only one annotation per genome is considered by any PanTools
functionality. When multiple annotations are included, the last added
annotation of a genome is automatically selected unless an
--annotations-file is included specifying which annotations to use.
This annotation file contains only annotation identifiers, each on a
separate line. The most recent annotation is used for genomes where no
annotation number is specified in the file. Below is an example where
the third annotation of genome 1 is selected and the second annotation
of genome 2 and 3.
1_3
2_2
3_2
Grouping proteins
Group
Generate homology groups based on similarity of protein sequences. The
resulting homology groups connect similar sequences in the pangenome
database. Homology groups contain not only orthologous pairs, but also
pairs of homologs duplicated after the speciation of the two species,
so-called in-paralogs. The sizes of the groups are controlled by the
--relaxation parameter that can be set very strict or more lenient,
depending on the evolutionary distance of the genomes. When you are
unsure which relaxation setting is most suitable for your dataset,
running the optimal_grouping
functionality is recommended.
Be aware that not every sequence within a homology group has to be similar to the other sequences. For example, two non-similar protein sequences each have a high-similarity hit with the same protein sequence but align to a different region, one at the start and one near the end of the sequence.
When you want to run group another time but with different parameters, the currently active grouping must first either be moved or removed. This can be achieved with the move- or remove_homology_groups functions.
--relaxation), the proteins are
connected with each other in the similarity graph. Every similarity
component is then passed to the MCL (Markov clustering) algorithm to
be possibly broken into several homology groups.Required software
Required arguments
--database-path/-dp Path to the pangenome database.
Optional arguments
--skip/-sk Exclude a selection of genomes.--reference/-ref Only include a selection of genomes.--threads/-tn The number of parallel working threads. Default
and minimum required threads is 3.--longest-transcript Only cluster the longest protein-coding
transcript of genes.--annotations-file/-af A text file with the identifiers of
annotations to be included, each on a separate line. The most recent
annotation is selected for genomes without an identifier.Optional arguments that influence the clustering sensitivity
--relaxation/-rn The relaxation in homology calls. Should be
in range [1-8], from strict to relaxed (default 1). IMPORTANT!
This argument automatically sets the four remaining arguments, stated
here below.--intersection-rate/-ir The fraction of k-mers that needs to
be shared by two intersecting proteins. Should be in range [0.001,
0.1] (default = 0.08).--similarity-threshold/-st The minimum normalized similarity
score of two proteins. Should be in range [1-99] (default = 95).--mcl-inflation/-mi The MCL inflation. Should be in range
[1-19] (default = 10.8).--contrast/-cn The contrast factor. Should be in range [0-10]
(default = 8).Example commands
$ pantools group -dp tomato_DB
$ pantools group -dp tomato_DB -tn 12 -rn 4
Output
pantools_homology_groups.txt, overview of the created homology groups. Each line represents one homology group, starting with the homology group (database) identifier followed by a colon (:) and mRNA identifiers (from GFF) that are separated by a space. To ensure all identifiers are unique in this file, the mRNA ids are extended by a hash symbol (#) and a genome number. The following line is example output of an homology group with two genes from genome 1 and 146:
14001754: DLACAPHP_00001_mRNA#1 OPJEMMMF_03822_mRNA#146
Relevant literature
Optimal grouping
Finding the most suitable settings for group
can be difficult and is always dependent on evolutionary distance of the
genomes in the pangenome. This functionality runs group on all eight
--relaxation settings, from strictest (d1) to the most relaxed (d8).
To find the optimal setting, complete and non-duplicated BUSCO genes
that are present in all genomes are used to validate each setting.
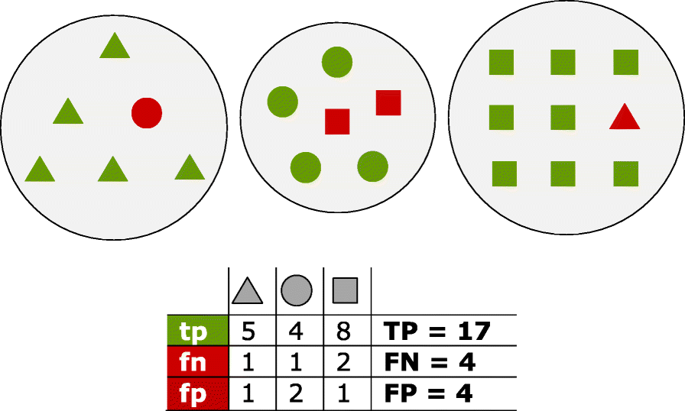
Fig. 1 Proteins of three distinct homology groups are represented as triangles, circles and squares. Green shapes are true positives (tp) which have been assigned to the true group; red shapes are false positives (fp) for the group they have been incorrectly assigned to, and false negatives (fn) for their true group
No grouping is active after running this function. Use the generated output files to identify a suitable grouping. Activate this grouping using change_grouping. An overview of the available groupings and used settings is stored in the ‘pangenome’ node (inside the database), or can be created by running grouping_overview.
Required software
Required arguments
--database-path/-dp Path to the pangenome database.--input-file/-if The output directory created by the
busco_protein function. This directory is
found inside the pangenome database, in the busco directory.Optional arguments
--skip/-sk Exclude a selection of genomes.--reference/-ref Only include a selection of genomes.--threads/tn Number of threads. The default and minimum
required threads is 3.--value Only consider a selection of relaxation settings (1-8
allowed).--fast Assume the optimal grouping is found when the F1-score
drops compared to the previous clustering round.--longest-transcript Only cluster protein sequences of the largest
transcript per gene.--annotations-file/-af A text file with the identifiers of
annotations to be included, each on a separate line. The most recent
annotation is selected for genomes without an identifier.Example commands
$ pantools optimal_grouping -dp bacteria_DB -if bacteria_DB/busco/bacteria_odb9
$ pantools optimal_grouping -dp bacteria_DB -if bacteria_DB/busco/bacteria_odb9 -tn 12 --fast
$ pantools optimal_grouping -dp bacteria_DB -if bacteria_DB/busco/bacteria_odb9 -tn 12 --fast --longest-transcript
$ pantools optimal_grouping -dp bacteria_DB -if bacteria_DB/busco/bacteria_odb9 -tn 12 --value 1,2,3,4
$ Rscript optimal_grouping.R
Output
After each clustering round, homology groups are incorporated in the graph. A text file with homology group and gene identifiers is stored in the group directory in the pangenome database. This file is named after the used sequence similarity threshold (25-95). Each line represents one homology group, starting with the homology group (database) identifier followed by a colon (:) and mRNA identifiers (from GFF) that are separated by a space. The mRNA identifiers are extended by a hash (#) and their genome number. The following line is example output of an homology group with two genes from genome 1 and 146:
14001754: DLACAPHP_00001_mRNA#1 OPJEMMMF_03822_mRNA#146
Output files are written to optimal_grouping directory inside the database.
grouping_overview.csv, a summary of the benchmark statistics. Use this file to find the most suitable grouping for your pangenome.
optimal_grouping.R, Rscript to plot FN and FP values per grouping.
counts_per_busco.info, a log file of the scoring. Shows in which homology groups the BUSCO genes were placed for the different groupings.
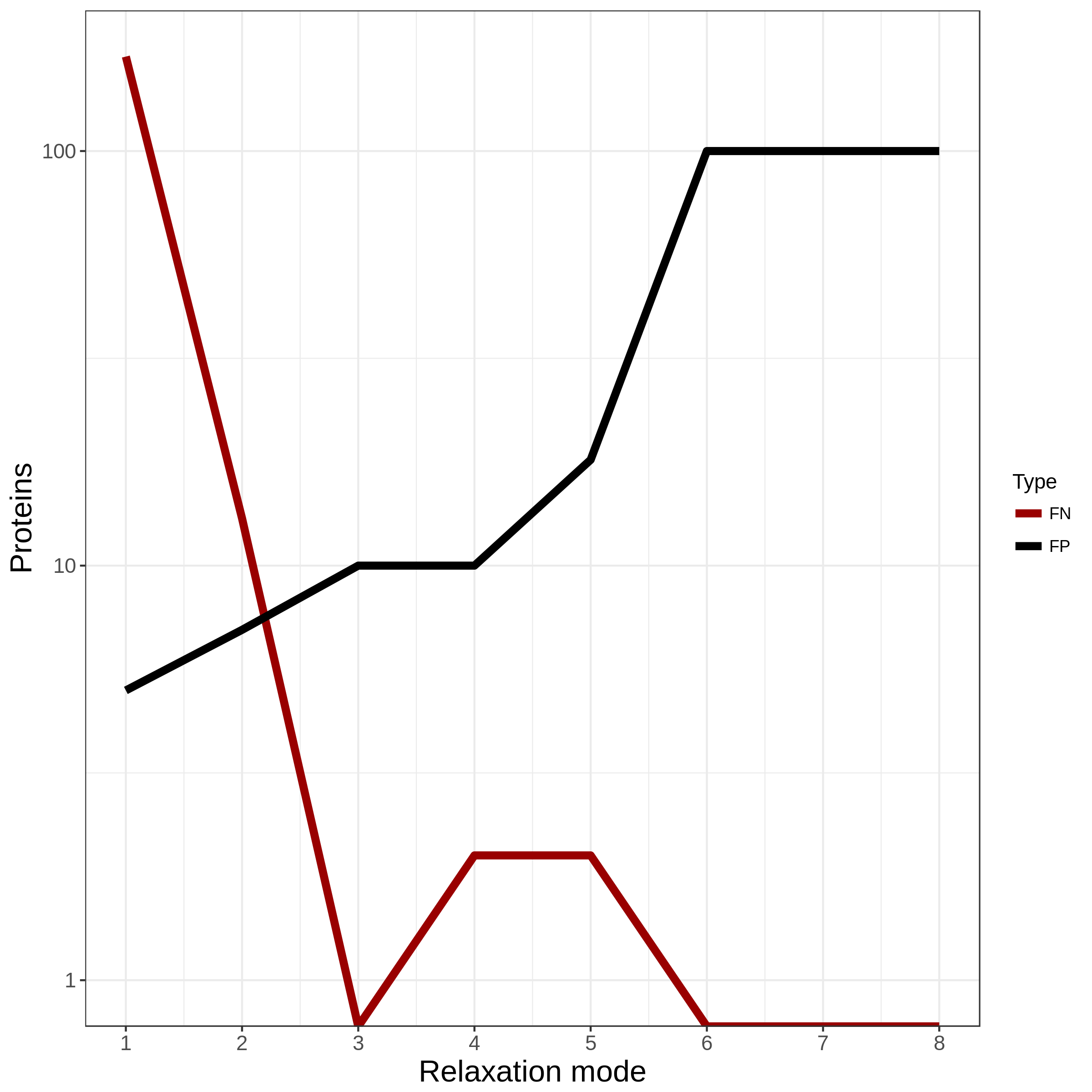
Fig. 2 :italic:`Example output of **optimal_grouping.R**. The number of FN and FP for all eight relaxation settings.`
Change grouping
Only a single homology grouping can be active in the pangenome. Use this function to change the active grouping version. Information of the available groupings and used settings is stored in the ‘pangenome’ node (inside the database) and can be created by running grouping_overview.
Required arguments
--database-path/-dp Path to the pangenome database.--version The version of homology grouping to become active.Example command
$ pantools change_grouping -dp tomato_DB --version 5
Build panproteome
Build a panproteome out of a set of proteins. By only including protein sequences, the usable functionalities are limited to a protein-based analysis, please see differences pangenome and panproteome. No additional proteins can be added to the panproteome, it needs to be rebuilt completely.
Required arguments
--database-path/-dp Path to the pangenome database.--proteomes-file/-pf A text file containing paths to FASTA
files of proteins to be added to the panproteome; each on a separate
line.Example input file
/always/proteins1.fasta
/use_the/proteins2.fasta
/full_path/proteins3.faa
Example command
$ pantools build_panproteome -dp proteome_DB -pf proteins.txt
Add genomes
Include additional genomes to an already available pangenome.
Required software
Required arguments
--database-path/-dp Path to the pangenome database.--genomes-file/-gf A text file containing paths to FASTA files
of genomes to be added to the pangenome; each on a separate line.Example input file
/use_the/genome4.fasta
/full_path/genome5.fasta
Example command
$ pantools add_genomes -dp pangenome_DB -gf extra_genomes.txt
Add phenotypes
Including phenotype data to the pangenome which allows the identification of phenotype specific genes, SNPs, functions, etc.. Altering the data is done by rerunning the command with an updated CSV file.
Values recognized as round number are converted to an Integer and to a Double when having one or multiple decimals.
Boolean types are identified by checking if the value matches ‘true’ or ‘false’, ignoring capitalization of letters.
String values remain completely unaltered except for spaces and quotes characters. Spaces are changed into an underscore (’_’) character and quotes are completely removed.
--value. For skewed data,
consider making the bins manually and include this as string
phenotype.Required arguments
--database-path/-dp Path to the pangenome database.--phenotype/-ph A CSV file containing the phenotype
information.Optional argument
--append Do not remove existing phenotype nodes but only add new
properties to it. If a property already exists, values from the new
file will overwrite the old.--value Number of bins used to group numerical values of a
phenotype.Example input file
The input file needs to be in .CSV format, a plain text file where each value is separated by a comma. The first row should start with ‘Genome,’ followed by the phenotype names and/or identifiers. The first column must start with genome numbers corresponding to the one in your pangenome. Phenotypes and metadata must be placed on the same line as their genome number. A field can remain empty when the phenotype for a genome is missing or unknown. Here below is an example of five genomes contains six phenotypes:
Genome,Gram,Region,Pathogenicity,Boolean,float,species
1,+,NL,3,True,0.1,Species
2,+,BE,,False,0.1,Species3
3,+,LUX,7,true,0.1,Species3
4,+,NL,9,false,0.1,Species3
5,+,BE,15,TRUE,0.1,Species1
Example command
$ pantools add_phenotype -dp tomato_DB --phenotype pheno.csv
$ pantools add_phenotype -dp tomato_DB -ph pheno.csv --append
Output
Phenotype information is stored in ‘phenotype’ nodes in the graph. An output file is written to the database directory.
phenotype_overview.txt, a summary of the available phenotypes in the pangenome
BUSCO
BUSCO attempts to provide a quantitative assessment of the completeness in terms of expected gene content of a genome assembly. Proteins are placed into categories of Complete and single-copy (S), Complete and duplicated (D), fragmented (F), or missing (M). This function is able to run BUSCO v3, v4 or v5 against protein sequences of the pangenome.
The number of reported duplicated genes in eukaryotes is often to high
as different protein isoforms are counted multiple times. To adjust the
imprecise duplication score, include the --longest-transcripts
argument to the command.
You don’t have a benchmark set?
When using BUSCO v3, go to https://busco.ezlab.org, download a odb9 set, and untar it with
tar -xvzf. Include the entire directory in the command using the--input-fileargument.For BUSCO v4 and v5, you only have to provide the odb10 database name with the
--input-fileargument, the database is downloaded automatically. To get a full list of the available datasets, runbusco --list-datasets.
Required software
BUSCO must be set to your $PATH. For v3, test if the
which run_BUSCO.py command displays the full path so it can accessed
anywhere. For v4 and v5, test if busco is executable.
Required arguments
--database-path/-dp Path to the pangenome database.--input-file/-if A BUSCO benchmark dataset.Optional arguments
--skip/-sk Exclude a selection of genomes.--reference/-ref Only include a selection of genomes.--name A string with questionable BUSCOs. Completeness (%) is
recalculated by excluding these genes.--version The BUSCO version. Select either ‘busco3’, ‘busco4’ or
‘busco5’ (default).--longest-transcript Only search against the longest
protein-coding transcript of genes.--annotations-file/-af A text file with the identifiers of
annotations to be included, each on a separate line. The most recent
annotation is selected for genomes without an identifier.Example commands
$ pantools busco_protein -dp bacteria_DB -if bacteria_odb10
$ pantools busco_protein -dp bacteria_DB -if busco_sets/bacteria_odb9/ --version busco3
$ pantools busco_protein -dp bacteria_DB -if busco_sets/bacteria_odb9/ --version busco3 --name POG093P01OY,POG093P0009,POG093P022K,POG093P027M,POG093P00Z2,POG093P013J
$ pantools busco_protein -dp bacteria_DB -if bacteria_odb10 --version busco4 --longest-transcript
Output
The BUSCO scores are stored inside BUSCO nodes of the pangenome graph. Output files are written to the busco directory inside the database.
busco_scores.txt, overview of the BUSCO scores per genome. Average and median statistics are calculated per category.
busco_overview.csv, a table which combines the completeness scores per genome together with the duplicated, fragmented and missing BUSCO genes.
hmm_overview.txt, a list of BUSCO genes showing the assigned categories per genome.
Add functional annotations
PanTools is able to incorporate functional annotations into the pangenome by reading output from various functional annotation tools.
Add functions
This function can integrate different functional annotations from a variety of annotation files. Currently available functional annotations: Gene Ontology, Pfam, InterPro, TIGRFAM, Phobius, SignalP and COG. The first time this function is executed, the Pfam, TIRGRAM, GO, and InterPro databases are integrated into the pangenome. Phobius, SignalP and COG annotations do not have separate nodes and are directly annotated on ‘mRNA’ nodes in the pangenome.
Gene names (or identifiers) from the input file are used to identify gene nodes in the pangenome. Only genes with an exactly matching name/identifier can be connected to functional annotation nodes! Use the same FASTA and GFF3 files that were used to construct the pangenome database.
Functional databases
Database versions in v3.4.0 repository
Version |
Download date (dd-mm-yyyy) |
|
|---|---|---|
Gene ontology |
2021-12-15 |
20-12-2021 |
Pfam |
35.0 |
20-12-2021 |
TIGRFAM |
15.0 |
01-10-2020 |
InterPro |
87+ |
Not included in repository |
We regularly check and update the four functional database. To update the functional database manually, download the following files and replace the old ones in the /pantools/addons/ directory. The TIGRFAM.info files are bundled in the TIGRFAMs_15.0_INFO.tar.gz file; download the file to addons/tigrfam and uncompress the tarball first. The first time running this function .INFO files are combined into a new file COMBINATION_INFO_FILES and removed afterwards.
File |
Database type |
Required directory |
Download link |
|---|---|---|---|
go.basic.obo |
GO |
addons |
|
gene_ontology.txt |
Pfam |
addons |
ftp://ftp.ebi.ac.uk/pub/databases/Pfam/releases//Pfam35.0/database_files/gene_ontology.txt.gz |
Pfam-A.clans.tsv |
Pfam |
addons |
ftp://ftp.ebi.ac.uk/pub/databases/Pfam/releases//Pfam35.0/Pfam-A.clans.tsv.gz |
interpro.xml |
InterPro |
addons |
https://ftp.ebi.ac.uk/pub/databases/interpro/current_release/interpro.xml.gz |
TIGRFAMS_GO_LINK |
TIGRFAM |
addons/tigrfam |
https://ftp.ncbi.nlm.nih.gov/hmm/TIGRFAMs/release_15.0/TIGRFAMS_GO_LINK |
TIGRFAMS_ROLE_LINK |
TIGRFAM |
addons/tigrfam |
https://ftp.ncbi.nlm.nih.gov/hmm/TIGRFAMs/release_15.0/TIGRFAMS_ROLE_LINK |
TIGR_ROLE_NAMES |
TIGRFAM |
addons/tigrfam |
https://ftp.ncbi.nlm.nih.gov/hmm/TIGRFAMs/release_15.0/TIGR_ROLE_NAMES |
TIGR00001.INFO to TIGR04571.INFO |
TIGRFAM |
addons/tigrfam |
https://ftp.ncbi.nlm.nih.gov/hmm/TIGRFAMs/release_15.0/TIGRFAMs_15.0_INFO.tar.gz |
Required arguments
--database-path/-dp Path to the pangenome database.--input-file/-if A text file with on each line a genome number
and the full path to the corresponding annotation file, separated by a
space.Optional arguments
--annotations-file/-af A text file with the identifiers of
annotations to be included, each on a separate line. The most recent
annotation is selected for genomes without an identifier.
Example command
$ pantools add_functions -dp tomato_DB -if f_annotations.txt
$ pantools add_functions -dp tomato_DB -if f_annotations.txt -af annotations.txt
Output
Functional annotations are incorporated in the graph. A log file is written to the log directory.
add_functional_annotations.log, a log file with the the number of added functions per type and the identifiers of functions that could not be included.
Example input files
The --input-file requires to be formatted like an annotation input
file. Each line of the file starts with the genome number followed by
the full path to an annotation file.
File type |
Recognized by pattern in file name |
|---|---|
InterProScan |
interpro & .gff |
eggNOG-mapper |
eggnog |
Phobius |
phobius |
SignalP |
signalp |
Custom file |
custom |
1 /mnt/scratch/interpro_results_genome_1.gff
1 /mnt/scratch/custom_annotation_1.txt
1 /mnt/scratch/phobius_1.txt
2 /mnt/scratch/signalp.txt
2 /mnt/scratch/eggnog_genome_2.annotations
2 /mnt/scratch/transmembrane_annotations.txt phobius
3 /mnt/scratch/ipro_results_genome_3.annot custom
Phobius and SignalP are not standard analyses of the InterProScan pipeline and require some additional steps during the InterProScan installation. Please take a look at our InterProScan install instruction to verify if the tools are part of the prediction pipeline. Phobius 1.01
Function type |
Allowed annotation file |
|---|---|
GO |
InterProscan .gff & custom annotation file |
Pfam |
InterProscan .gff & custom annotation file |
InterPro |
InterProscan .gff & custom annotation file |
TIGRFAM |
InterProscan .gff & custom annotation file |
Phobius |
InterProscan .gff & Phobius 1.01 output |
SignalP |
InterProscan .gff, signalP 4.1 output, signalP 5.0 output |
COG |
eggNOG-mapper |
InterProScan gff file:
##gff-version 3
##interproscan-version 5.52-86.0
AT4G21230.1 ProSiteProfiles protein_match 333 620 39.000664 + . date=06-10-2021;Target=mRNA.AT4G21230.1 333 620;Ontology_term="GO:0004672","GO:0005524","GO:0006468";ID=match$42_333_620;signature_desc=Protein kinase domain profile.;Name=PS50011;status=T;Dbxref="InterPro:IPR000719"
AT3G08980.5 TIGRFAM protein_match 25 101 3.7E-14 + . date=06-10-2021;Target=mRNA.AT3G08980.5 25 101;Ontology_term="GO:0006508","GO:0008236","GO:0016020";ID=match$66_25_101;signature_desc=sigpep_I_bact: signal peptidase I;Name=TIGR02227;status=T;Dbxref="InterPro:IPR000223"
AT2G17780.2 Phobius protein_match 338 354 . + . date=06-10-2021;Target=AT2G17780.2 338 354;ID=match$141_338_354;signature_desc=Region of a membrane-bound protein predicted to be embedded in the membrane.;Name=TRANSMEMBRANE;status=T
AT2G17780.2 Phobius protein_match 1 337 . + . date=06-10-2021;Target=AT2G17780.2 1 337;ID=match$142_1_337;signature_desc=Region of a membrane-bound protein predicted to be outside the membrane, in the extracellular region.;Name=NON_CYTOPLASMIC_DOMAIN;status=T
AT3G11780.2 SignalP_EUK protein_match 1 24 . + . date=06-10-2021;Target=mRNA.AT3G11780.2 1 24;ID=match$230_1_24;Name=SignalP-noTM;status=T
AT1G04300.2 CDD protein_match 40 114 1.54717E-13 + . date=06-10-2021;Target=mRNA.AT1G04300.2 40 114;Ontology_term="GO:0005515";ID=match$212_40_114;signature_desc=MATH;Name=cd00121;status=T;Dbxref="InterPro:IPR002083"
eggNOG-mapper (tab separated) file:
#query_name seed_eggNOG_ortholog seed_ortholog_evalue seed_ortholog_score best_tax_level Preferred_name GOs EC KEGG_ko KEGG_Pathway KEGG_Module KEGG_Reaction KEGG_rclass BRITE KEGG_TC CAZy BiGG_Reaction taxonomic scope eggNOG OGs best eggNOG OG COG Functional cat. eggNOG free text desc.
ATKYO-2G54530.1 3702.AT2G35130.2 1.9e-179 636.0 Brassicales GO:0003674,GO:0003676,GO:0003723,GO:0003824,GO:0004518,GO:0004519,GO:0005488,GO:0005575,GO:0005622,GO:0005623,GO:0006139,GO:0006725,GO:0006807,GO:0008150,GO:0008152,GO:0009451,GO:0009987,GO:0016070,GO:0016787,GO:0016788,GO:0034641,GO:0043170,GO:0043226,GO:0043227,GO:0043229,GO:0043231,GO:0043412,GO:0044237,GO:0044238,GO:0044424,GO:0044464,GO:0046483,GO:0071704,GO:0090304,GO:0090305,GO:0097159,GO:1901360,GO:1901363 Viridiplantae 37R67@33090,3GAUT@35493,3HNDD@3699,KOG4197@1,KOG4197@2759 NA|NA|NA E Pentacotripeptide-repeat region of PRORP
ATKYO-UG22500.1 3712.Bo02269s010.1 7.5e-35 153.7 Brassicales Viridiplantae 29I9W@1,2RRH4@2759,383W6@33090,3GWQZ@35493,3I1A9@3699 NA|NA|NA
ATKYO-1G60060.1 3702.AT1G48090.1 0.0 6241.0 Brassicales ko:K19525 ko00000 Viridiplantae 37IJB@33090,3GAN0@35493,3HQ90@3699,COG5043@1,KOG1809@2759 NA|NA|NA U Vacuolar protein sorting-associated protein
ATKYO-3G74720.1 3702.AT3G52120.1 7.2e-245 852.8 Brassicales ko:K13096 ko00000,ko03041 Viridiplantae 37QYY@33090,3G9VU@35493,3HRDK@3699,KOG0965@1,KOG0965@2759 NA|NA|NA L SWAP (Suppressor-of-White-APricot) surp domain-containing protein D111 G-patch domain-containing protein
ATKYO-4G41660.1 3702.AT4G16340.1 0.0 3392.1 Brassicales GO:0003674,GO:0005085,GO:0005088,GO:0005089,GO:0005488,GO:0005515,GO:0005575,GO:0005622,GO:0005623,GO:0005634,GO:0005737,GO:0005783,GO:0005829,GO:0005886,GO:0006810,GO:0008064,GO:0008150,GO:0008360,GO:0009605,GO:0009606,GO:0009628,GO:0009629,GO:0009630,GO:0009958,GO:0009966,GO:0009987,GO:0010646,GO:0010928,GO:0012505,GO:0016020,GO:0016043,GO:0016192,GO:0017016,GO:0017048,GO:0019898,GO:0019899,GO:0022603,GO:0022604,GO:0023051,GO:0030832,GO:0031267,GO:0032535,GO:0032956,GO:0032970,GO:0033043,GO:0043226,GO:0043227,GO:0043229,GO:0043231,GO:0044422,GO:0044424,GO:0044425,GO:0044432,GO:0044444,GO:0044446,GO:0044464,GO:0048583,GO:0050789,GO:0050793,GO:0050794,GO:0050896,GO:0051020,GO:0051128,GO:0051179,GO:0051234,GO:0051493,GO:0065007,GO:0065008,GO:0065009,GO:0070971,GO:0071840,GO:0071944,GO:0090066,GO:0098772,GO:0110053,GO:1902903 ko:K21852 ko00000,ko04131 Viridiplantae 37QIM@33090,3G8RK@35493,3HSFN@3699,KOG1997@1,KOG1997@2759 NA|NA|NA T Belongs to the DOCK family
A custom input file must consist of two tab or comma separated columns. The first column should contain a gene/mRNA id, the second an identifier from one of four functional annotation databases: GO, Pfam, InterPro or TIGRFAM.
AT5G23090.4,GO:0046982
AT5G23090.4,IPR009072
AT1G27540.2,PF03478
AT2G18450.1,TIGR01816
Phobius 1.01 ‘short’ (tab separated) input file:
SEQENCE ID TM SP PREDICTION
mRNA-YPR204W 0 0 o
mRNA-ndhB-2_1 6 Y n5-16c21/22o37-57i64-83o89-113i134-156o168-189i223-246o
Phobius 1.01 ‘long’ (tab separated) input file:
ID mRNA-YPR204W
FT DOMAIN 1 1032 NON CYTOPLASMIC.
//
ID mRNA-ndhB-2_1
FT SIGNAL 1 21
FT DOMAIN 1 4 N-REGION.
FT DOMAIN 5 16 H-REGION.
FT DOMAIN 17 21 C-REGION.
FT DOMAIN 22 36 NON CYTOPLASMIC.
FT TRANSMEM 37 57
FT DOMAIN 58 63 CYTOPLASMIC.
FT TRANSMEM 64 83
FT DOMAIN 84 88 NON CYTOPLASMIC.
FT TRANSMEM 89 113
FT DOMAIN 114 133 CYTOPLASMIC.
FT TRANSMEM 134 156
FT DOMAIN 157 167 NON CYTOPLASMIC.
FT TRANSMEM 168 189
FT DOMAIN 190 222 CYTOPLASMIC.
FT TRANSMEM 223 246
FT DOMAIN 247 253 NON CYTOPLASMIC.
//
SignalP 4.1 ‘short’ (tab separated) input file:
# name Cmax pos Ymax pos Smax pos Smean D ? Dmaxcut Networks-used
mRNA-rpl2-3 0.148 20 0.136 20 0.146 3 0.126 0.131 N 0.450 SignalP-noTM
mRNA-cox2 0.107 25 0.132 12 0.270 4 0.162 0.148 N 0.450 SignalP-noTM
mRNA-cox2_1 0.850 17 0.776 17 0.785 2 0.717 0.753 Y 0.500 SignalP-TM
SignalP 5.0 ‘short’ (tab separated) input file:
# SignalP-5.0 Organism: Eukarya Timestamp: 20211122233246
# ID Prediction SP(Sec/SPI) OTHER CS Position
AT3G26880.1 SP(Sec/SPI) 0.998803 0.001197 CS pos: 21-22. VYG-KK. Pr: 0.9807
mRNA-rpl2-3 OTHER 0.001227 0.998773
Relevant literature
Add antiSMASH gene clusters
Read antiSMASH output and incorporate Biosynthetic Gene Clusters (BGC) nodes into the pangenome database. A ‘bgc’ node holds the gene cluster product, the cluster address and has a relationship to all gene nodes of the cluster. For this function to work, antiSMASH should be performed with the same FASTA and GFF3 files used for building the pangenome. antiSMASH output will not match the identifiers of the pangenome when no GFF file was included.
As of PanTools v3.3.4 the required antiSMASH version is 6.0.0. Gene cluster information is parsed from the .JSON file that is generated in each run. We try to keep the parser updated with newer versions but please contact us when this is no longer the case.
Version |
Version date |
|
|---|---|---|
antiSMASH |
6.0.0 |
21-02-2021 |
Required arguments
--database-path/-dp Path to the pangenome database.--input-file/-if A text file with on each line a genome number
and the full path to the corresponding antiSMASH output file,
separated by a space.Optional arguments
--annotations-file/-af A text file with the identifiers of
annotations to be included, each on a separate line. The most recent
annotation is selected for genomes without an identifier.
Example input file
The --input-file requires to be formatted like a regular annotation
input file. Each line of the file starts with the genome number followed
by the full path to the JSON file.
1 /mnt/scratch/IPO3844/antismash/IPO3844.json
4 /home/user/IPO3845/antismash/IPO3845.json
Example command
$ pantools add_antismash -dp tomato_DB -if clusters.txt
Removing data
The following functionalities allow the removal of large sets of nodes and relationships from the pangenome. These functions will first ask for a confirmation before the nodes are actually removed. Be careful, the data is not backed up and removing nodes or properties means it is permanently gone.
Remove nodes
Remove a selection of nodes and their relationships from the pangenome. For a pangenome database the following nodes cannot be removed: nucleotide, pangenome, genome, sequence. When using a panproteome, mRNA nodes cannot be removed.
Required argument
--database-path/-dp Path to the pangenome database.
Requires either one of the following arguments
--node one or multiple node identifiers, separated by a comma.--label a node label, all nodes matching the label are removed.Optional arguments
Both optional arguments can only be used in combination with
--label.
--skip/-sk Do not remove nodes of the selected genomes.--reference/-ref Only remove nodes of the selected genomes.Example commands
$ pantools remove_nodes -dp tomato_DB --node 10348734,10348735,10348736
$ pantools remove_nodes -dp tomato_DB --label pfam
$ pantools remove_nodes -dp tomato_DB --label interpro --reference 2-6
Remove phenotypes
Delete phenotype nodes or remove specific phenotype information from
the nodes. The specific phenotype property needs to be specified with
--phenotype. When this argument is not included, phenotype nodes
are removed.
Required argument
--database-path/-dp Path to the pangenome database.
Optional arguments
--phenotype/-ph name of the phenotype. All information of the
given phenotype is removed from ‘phenotype’ nodes.--skip/-sk Do not remove nodes of the selected genomes.--reference/-ref Only remove nodes of the selected genomes.Example commands
$ pantools remove_phenotype -dp tomato_DB
$ pantools remove_phenotype -dp tomato_DB --phenotype color
$ pantools remove_phenotype -dp tomato_DB --phenotype color --skip 11,12
Remove annotations
Remove all the genomic features that belong to annotations, such as gene, mRNA, exon, tRNA, and feature nodes. Functional annotation nodes are not removed with this function but can be removed with remove_nodes. Removing annotations can be done in two ways:
Selecting genomes with
--referenceor--skip, for which all annotation features will be removed.Remove specific annotations by providing a text file with identifiers via the
--annotations-fileargument.
Required argument
--database-path/-dp Path to the pangenome database.
Requires either one of the following arguments
--skip/-sk a selection of genomes excluded from the removal of
annotations.--reference/-ref a selection of genomes for which all
annotations will be removed.--annotations-file/-af A text file with the identifiers of
annotations to be removed, each on a separate line.Example input file
The input file should be a single line with annotation identifiers separated by a comma. The following example will remove the first annotations of genome 1, 2 and 3 and the second annotation of genome 1.
1_1
1_2
2_1
3_1
Example command
$ pantools remove_annotations --skip 3,4,5
$ pantools remove_annotations -af annotations.txt
Move or remove grouping
As only one grouping can be active at the time, the currently active grouping needs to be removed or inactivated before group can be run again.
remove_grouping deletes all ‘homology_group’ nodes and ‘is_similar’ relations between ‘mRNA’ nodes from the database.
move_grouping relabels ‘homology_group’ nodes to ‘inactive_homology_group’. The moved grouping can be activated again with change_grouping.
Required argument
--database-path/-dp Path to the pangenome database.
Optional arguments for remove_grouping
--version Select a specific grouping version to be removed. Two
additional options: ‘all’ to remove all groupings and ‘all_inactive’ to
remove all inactive groupings.--fast Do not remove the ‘is_similar’ relationships between mRNA
nodes. This does not influence the next grouping.Example command
$ pantools move_grouping -dp tomato_DB
$ pantools remove_grouping -dp tomato_DB
$ pantools remove_grouping -dp tomato_DB --version 1
$ pantools remove_grouping -dp tomato_DB --version all --fast
$ pantools remove_grouping -dp tomato_DB --version all_inactive